
by Rostam Golesorkhtabar, Pasquale Pavone, & Konstantin Lion for exciting oxygen
Purpose: In this tutorial you will learn how to set up and execute exciting calculations using the ElaStic@exciting interface, which allows to obtain full elastic tensors (elastic constants) of crystal systems for any crystal structure. In addition, the application of ElaStic@exciting to the determination of the elastic constants of C in the diamond phase and of hexagonal Be is explicitly presented.
|
Table of Contents
|
0. Define relevant environment variables
Read the following paragraphs before starting with the rest of this tutorial!
Before starting, be sure that relevant environment variables are already defined as specified in How to set environment variables for tutorials scripts. Here is a list of the scripts which are relevant for this tutorial with a short description.
- ElaStic@exciting-setup.py: Python script for generating structures at different strains.
- ElaStic@exciting-submit.sh: (Bash) shell script for running a series of exciting calculations.
- ElaStic@exciting-analyze.py: Python script for fitting the energy-vs-strain and CVe-vs-strain curves.
- ElaStic@exciting-clean.sh: (Bash) shell script for cleaning unnecessary files.
- ElaStic@exciting-result.py: Python script for calculating elastic constants.
- ElaStic_Result_Energy_2nd.py: Python script for calculating second-order elastic constants.
- ElaStic_Result_Energy_3rd.py: Python script for calculating third-order elastic constants.
- ElaStic_xyz2XYZ.py: Python script for the conversion of second-order elastic constants to different coordinate systems.
- exciting2sgroup.py: Python script for converting the exciting input to an sgroup input file.
- Grace.par: File containing xmgrace parameters needed by visualization tools.
Requirements: Bash shell. Python numpy, lxml, math, sys and os libraries. xmgrace (visualization tool).
From now on the symbol $ will indicate the shell prompt.
Extra requirement: Tool for space-group determination
The scripts in this tutorial use the sgroup tool. If you have not done before, this tool should be downloaded and installed.
1. Set up the calculations
i) Preparation of the input file
The first step is to create a directory for each system that you want to investigate. In this tutorial, we first consider as an example carbon in the diamond structure (the procedure we use is, nevertheless, valid for any system). Thus, we will create a directory C_2nd and we move inside it.
$ mkdir C_2nd
$ cd C_2ndInside this directory, we create or copy an optimized structure file for diamond named input.xml. This file could look like the following.
<input> <title>Diamond: 2nd-order elastic constants</title> <structure speciespath="$EXCITINGROOT/species"> <crystal scale="6.7165"> <basevect> 0.5 0.5 0.0 </basevect> <basevect> 0.5 0.0 0.5 </basevect> <basevect> 0.0 0.5 0.5 </basevect> </crystal> <species speciesfile="C.xml" rmt="1.30"> <atom coord="0.00 0.00 0.00"/> <atom coord="0.25 0.25 0.25"/> </species> </structure> <groundstate xctype="GGA_PBE_SOL" ngridk="6 6 6" swidth="0.0001" gmaxvr="18" maxscl="30"> </groundstate> <relax/> </input>
To the purposes of this tutorial, this file can be saved with any name. However, notice that the input file for a direct exciting calculation must be always called input.xml.
Be sure to set the correct path for the exciting root directory (indicated in this example by $EXCITINGROOT) to the one pointing to the place where the exciting directory is placed. In order to do this, use the command
$ SETUP-excitingroot.shii) Generation of input files for distorted structures
All strains considered in this tutorial are Lagrangian strains.
In order to generate input files for a series of distorted structures, you have to run the script ElaStic@exciting-setup.py, which will produce the following output on the screen.
$ ElaStic@exciting-setup.py
2nd ---=> 2
3rd ---=> 3
>>>> Please choose the order of the elastic constant (choose '2' or '3'): 2
>>>> Please enter the exciting input file name: input.xml
Number and name of space group: 227 (F d -3 m) [origin choice 2]
Cubic I structure in the Laue classification.
This structure has 3 independent second-order elastic constants.
>>>> Please enter the maximum Lagrangian strain
The suggested value is between 0.030 and 0.150: 0.05
The maximum Lagrangian strain is 0.05
>>>> Please enter the number of the distorted structures [odd number > 4]: 11
The number of the distorted structures is 11
$Entry values must be typed on the screen when requested. In this case, the entries are the following.
- 2, the order of the elastic constants in which you are interested.
- input.xml, the name of the input file.
- 0.05, the absolute value of the maximum strain for which we want to perform the calculation.
- 11, the number of deformed structures equally spaced in strain, which are generated between the maximum negative strain and the maximum positive one.
The script generates some new directories and files, which you can list using the following command (in the C_2nd directory).
$ ls
input.xml Distorted_Parameters Dst01 Dst02 Dst03 INFO_ElaStic Structures_exciting sgroup.out
$You may also list the single directories, e.g., if you list Dst01 you will see the following (sub)directories.
$ ls Dst01
Dst01_01 Dst01_02 Dst01_03 Dst01_04 Dst01_05 Dst01_06 Dst01_07 Dst01_08 Dst01_09 Dst01_10 Dst01_11
$3. Execute the calculations
To execute the series of calculations, run (in the C_2nd directory) the script ElaStic@exciting-submit.sh, which will produce an output on the screen similar to the following.
$ ElaStic@exciting-submit.sh
+--------------------------------------+
| SCF calculation of "Dst01_01" starts |
+--------------------------------------+
Elapsed time = 0m8s
+--------------------------------------+
| SCF calculation of "Dst01_02" starts |
+--------------------------------------+
Elapsed time = 0m8s
+--------------------------------------+
| SCF calculation of "Dst01_03" starts |
+--------------------------------------+
...
+--------------------------------------+
| SCF calculation of "Dst03_11" starts |
+--------------------------------------+
Elapsed time = 0m40s
$After the complete run, elastic constants can be calculated.
The running time necessary for performing a complete calculation of the elastic constants of diamond using computational parameters as included in the above file input.xml, is less than one hour (depending on the machine processor you are using).
4. Analyzing the calculations
Elastic constants are obtained from energy calculations using a similar method to the one illustrated in Energy vs. strain calculations. Within this approach, second derivatives of the energy curves are evaluated with the help of a curve fitting. Calculations performed above were producing 11 points for each energy curve.
Now, in order to calculate elastic constants, we have to analyze our calculations. The script ElaStic@exciting-analyze.py allows the analysis of the dependence of the calculated derivatives of the energy-vs-strain curve on
- the range of distortions included in the fitting procedure (the x axis in xmgrace plots),
- the degree of the polynomial fit used in the fitting procedure (different color curves in xmgrace plots).
The ElaStic@exciting-analyze.py script is executed as follows.
$ ElaStic@exciting-analyze.pyAt this point, three xmgrace plots will appear on your screen automatically (for more information on how to deal with xmgrace plots, see Xmgrace: A Quickstart). The plots should be similar to the ones displayed in the following.
- Results for the distortion Dst01

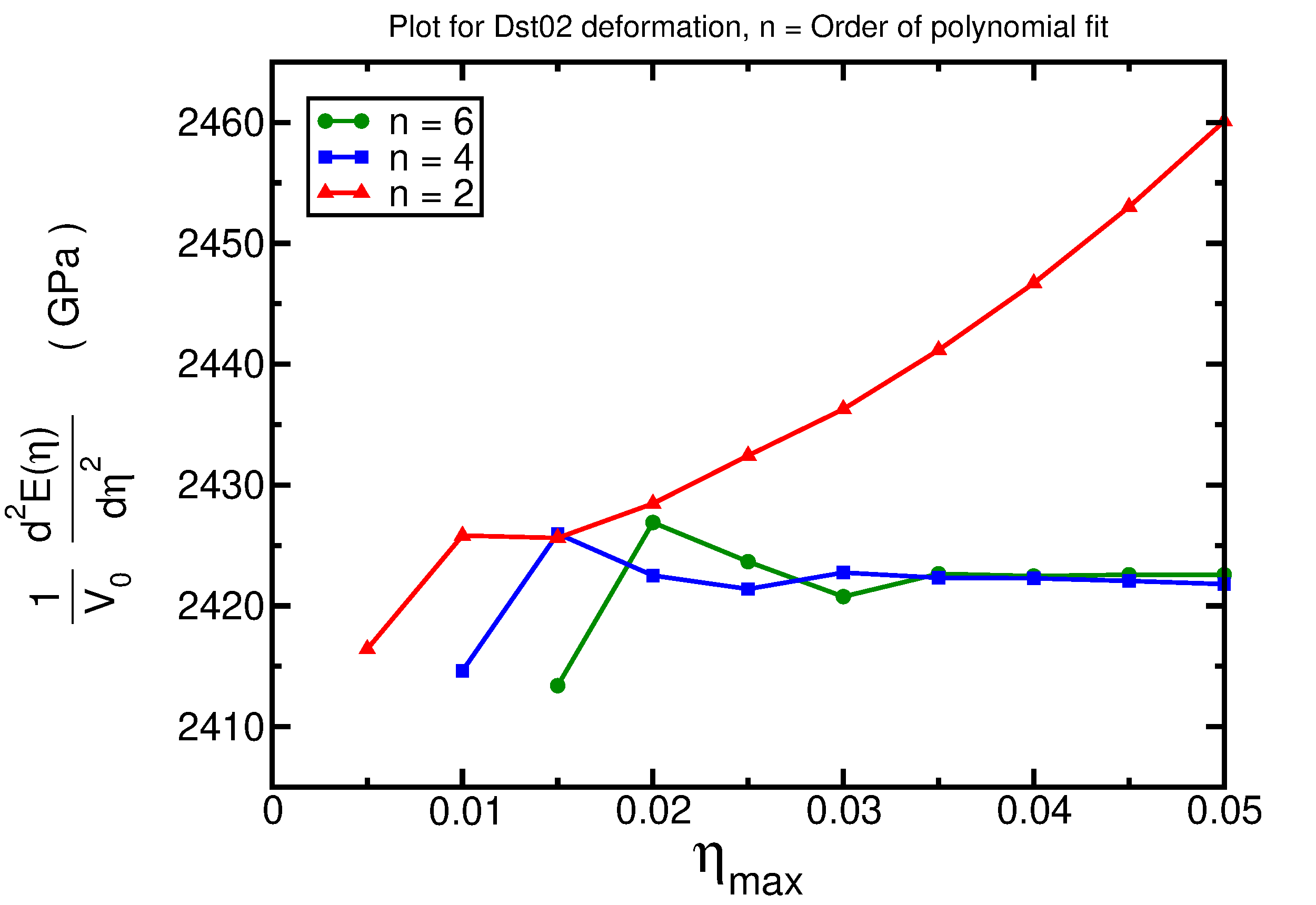
- Results for the distortion Dst02

- Results for the distortion Dst03

The previous plots can be used to determine the best range of deformations and order of polynomial fit for each distortion.
As an example, we analyze the first plot, corresponding to the distortion Dst01. Distortion types are listed in the file Distorted_Parameter. By examining this file, we can see that the Dst01 distortion corresponds to an applied Lagrangian strain in the Voigt notation with the form (η,η,η,0,0,0), where η is a strain parameter. This deformation type is directly connected with the bulk module B0. For each distortion type, a plot appears. This plot contains the second derivative of the energy with respect to the strain parameter η as a function of the maximum strain and of the order of polynomial fit. By analyzing the plot for the Dst01 distortion, we notice that curves corresponding to the higher order of the polynomial used in the fit show a horizontal plateau at about 4040-4045 GPa. This can be assumed to be the optimal value for the second derivative, from the point of view of the fit and corresponding to the choice of the other computational parameters (further information on this topic can be found here). For this distortion type, this value equals 9 times the bulk modulus. Thus, the extracted value of the bulk modulus is about 449 GPa.
As we have mentioned before many times, the quality of the results strongly depends on the numerical parameters used for the calculation. A better converged result (using a 10$\times$10$\times$10 k-grid, rgkmax = "8.0", and 21 deformed structures) can be found below.


5. Numerical values of the elastic constants
The script ElaStic@exciting-analyze.py used in the previous section, generates the file ElaStic_2nd.in, which will be discussed here. In order to obtain the numerical values of the second-order elastic constants of diamond, you should use the following procedure. The first step is to edit the file ElaStic_2nd.in, which should have the form
Dst01 eta_max Fit_order
Dst02 eta_max Fit_order
Dst03 eta_max Fit_orderIn each row of this file, you should insert a value for eta_max and Fit_order. According to the result of the visual analisys of the previous figures, these two values are extracted by considering the plateau regions of the corresponding plot: eta_max is a value of maximum distortion located in the plateau region of the curve representing the polynomial fit of order Fit_order. In our case, we can choose the parameters as listed in the example ElaStic_2nd.in below
Dst01 0.05 6
Dst02 0.05 6
Dst03 0.05 6Now, in order to calculate elastic constants and moduli, you should run the ElaStic@exciting-result.py script.
$ ElaStic@exciting-result.pyFinally, numerical values of the elastic constants for diamond are written in the output file ElaStic_2nd.out.
Experimental reference elastic constants for diamond:
- C11 = 1076 GPa
- C12 = 125 GPa
- C44 = 577 GPa
- B0 = 452 GPa
6. Set up the calculation for Be
In the next example, we consider the calculation of the elastic constants of hexagonal Be.
First, create a new directory Be_2nd and move inside it.
$ mkdir Be_2nd
$ cd Be_2ndInside this directory, create the file input.xml corresponding to the optimized input file for the equilibrium structure of Be.
<input> <title>Be: 2nd-order elastic constants</title> <structure speciespath="$EXCITINGROOT/species"> <crystal scale="4.3207"> <basevect> 1.00000000 0.00000000 0.00000000 </basevect> <basevect> -0.50000000 0.86602540 0.00000000 </basevect> <basevect> 0.00000000 0.00000000 1.52257736 </basevect> </crystal> <species speciesfile="Be.xml" rmt="1.85"> <atom coord="0.66666667 0.33333333 0.75000000"/> <atom coord="0.33333333 0.66666667 0.25000000"/> </species> </structure> <groundstate xctype="GGA_PBE_SOL" ngridk="6 6 4" rgkmax="6.5" maxscl="30"> </groundstate> <relax/> </input>
Do not forget to update the string "$EXCITINGROOT" inside the file input.xml.
Then, we generate a set of exciting input files with the script ElaStic@exciting-setup.py.
$ ElaStic@exciting-setup.py
2nd ---=> 2
3rd ---=> 3
>>>> Please choose the order of the elastic constant (choose '2' or '3'): 2
>>>> Please enter the exciting input file name: input.xml
Number and name of space group: 194 (P 63/m m c)
Hexagonal I structure in the Laue classification.
This structure has 5 independent second-order elastic constants.
>>>> Please enter the maximum Lagrangian strain
The suggested value is between 0.030 and 0.150: 0.05
The maximum Lagrangian strain is 0.05
>>>> Please enter the number of the distorted structures [odd number > 4]: 21
The number of the distorted structures is 21
$From now on, the procedure is completely equivalent to the one used for diamond. The analysis of the result can be made as in the previous example. Please, notice that the numerical parameters used in this example do not guarantee full convergence of results.